
Genomic coverage of retrotransposons in pig
The landscape of mobilome in pig was annotated using the RepeatMasker program using a custom library (click to download file 3-Repbase-pig-mobileDNAname-fl-20220422), including the de novo identified elements and known repeats from the Repbase and Dfam databases. The data demonstrate that the genomic coverage of mobilome in the pig is generally similar to that of the majority of mammals. Four types of TE (LINE, LTR, SINE, and DNA) accounted for 40.72% (∼1019 Mb) of the pig genome, with retrotransposons representing the vast majority, at 37.13% of the genome (∼929.1 Mb), with L1s have the highest genomic coverage, followed by SINEs and LTRs, whereas DNA TEs, mainly representing by hAT and Tc1/mariner superfamilies with extinct activities, accounted for the minority, at 1.99% (∼50.6 Mb) (Fig.1).
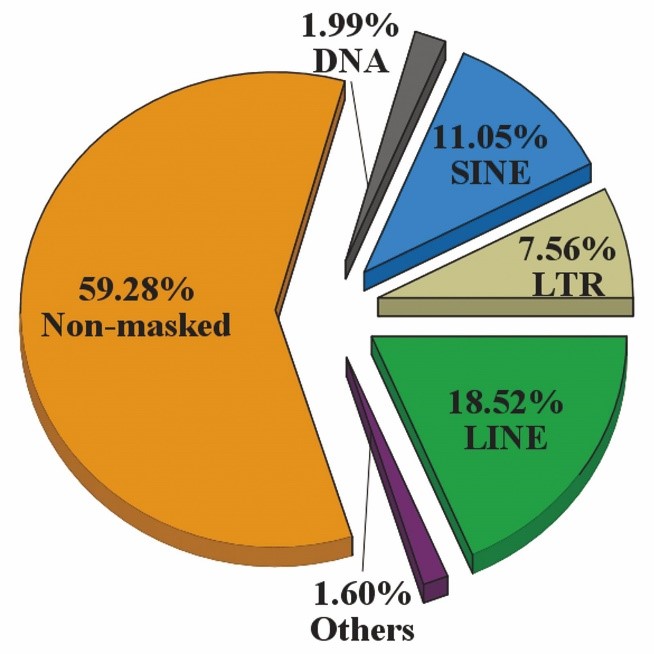
Fig. 1 Genomic coverage of retrotransposon types (LINEs, LTRs, SINEs) in the pig genome.
Reference: Chen C, Wang W, Wang X, Shen D, Wang S, Wang Y, Gao B, Wimmers K, Mao J, Li K, Song C. Retrotransposons evolution and impact on lncRNA and protein coding genes in pigs. Mob DNA. 2019 May 6;10:19. doi: 10.1186/s13100-019-0161-8. PMID: 31080521; PMCID: PMC6501411.
Retrotransposon classification in pig
De novo annotation of the genome of pig revealed that there are three main clades of retrotransposons: (1) L1s, which are classified into four distinct families and 51 distinct subfamilies, and about 100 putatively active L1 elements are identified in the pig genome, with a length between 7 kb and 9 kb, they contain a 5′UTR with length ranging from 1.5 kb to 3.2 kb, a 3′UTR of about 270 bp, two open-reading frames (296 aa ORF1 and 1272 aa ORF2), and a relatively long (about 520 bp) IGR that separates the two ORFs. L1 insertions typically end with an A-rich tail and are flanked by short (< 20 bp) target site duplication (Fig. 2A); (2) SINEs, which were represented by three distinct families and 25 subfamilies, and All SINE elements of SINEA, SINEB, and SINEC families showed similar structure organization, with a tRNA head, a TC-rich region, a GC-rich region, and an A-rich tail (Fig. 2B); (3) ERVs, which were classified into 18 families and found two most “modern” subfamilies in the pig genome. Most ERVs were typically between 8.5 Kb and 11 Kb in length, and the length of LTRs varied from 110 to 702 bp. Each of the two youngest subfamilies of ERVs (ERV6A and ERV6B) contained one putatively active ERV element with lengths of 8918 bp (chr5:92185133–92194050 -) and 8757 bp (chr9:138895584–138904340 -), respectively. The putatively active ERV element of ERV6A encoded a 1, 748 aa peptides containing gag, pol, and env, which are essential for replication, and flanked with 702 bp LTRs, while the active ERV of ERV6B subfamily encoded an 1, 776 aa peptide harboring gag, pol, and env, but flanked with 629 bp LTRs (Fig. 2C)
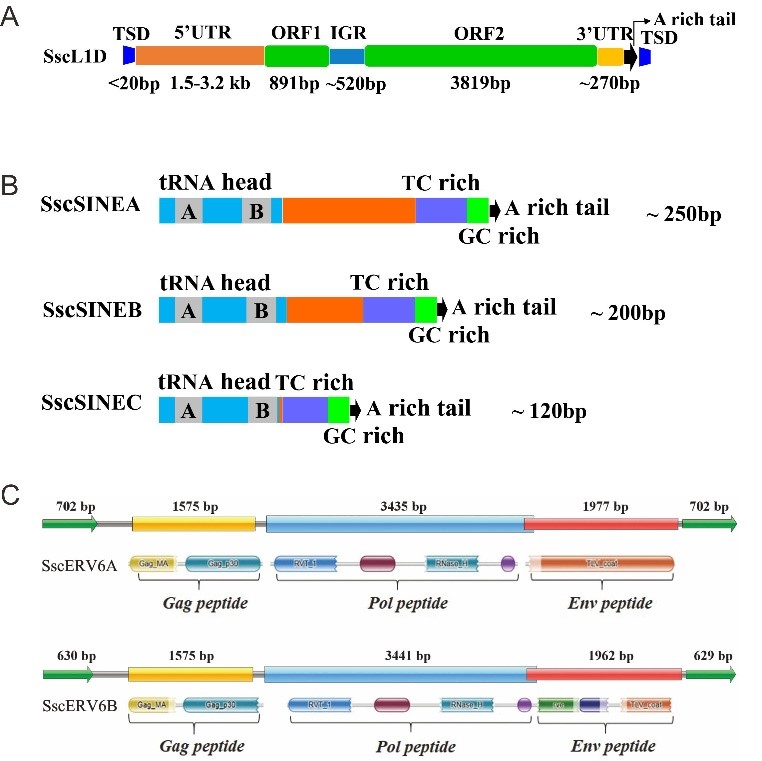
Fig 2. Structural schematics of L1, SINE and ERV in pig genome. (A), Structural schematics of the putatively active L1s. (B), Structural schematics of pig-specific SINE families (SINEA, SINEB, andSINEC). (C), Structural schematics of the ERV6A and ERV6B.
Reference: Chen C, Wang W, Wang X, Shen D, Wang S, Wang Y, Gao B, Wimmers K, Mao J, Li K, Song C. Retrotransposons evolution and impact on lncRNA and protein coding genes in pigs. Mob DNA. 2019 May 6;10:19. doi: 10.1186/s13100-019-0161-8. PMID: 31080521; PMCID: PMC6501411.
SINE RIPs mined from assembled genome data
Molecular markers based on retrotransposon insertion polymorphisms (RIPs) have been developed and are widely used in plants and animals. There are a large number of structural variations mediated by retrotransposons in the pig genome. Our analysis revealed that SINEA1–3 elements, particularly SINEA1, are high polymorphic across different pig breeds. Then we developed a genome-wide SINE RIP mining protocol and obtained 36,284 SINE RIPs, with over 80 % accuracy and an even distribution in chromosomes (14.5/Mb, Fig.4). Over 65 % of pig SINE RIPs overlap with genes, most of them (> 95 %) are in introns. Nearly half of the RIPs are common in these pig breeds. Sixteen SINE RIPs were applied for population genetic analysis in 23 pig breeds, the phylogeny tree and cluster analysis were generally consistent with the geographical distributions of native pig breeds in China.
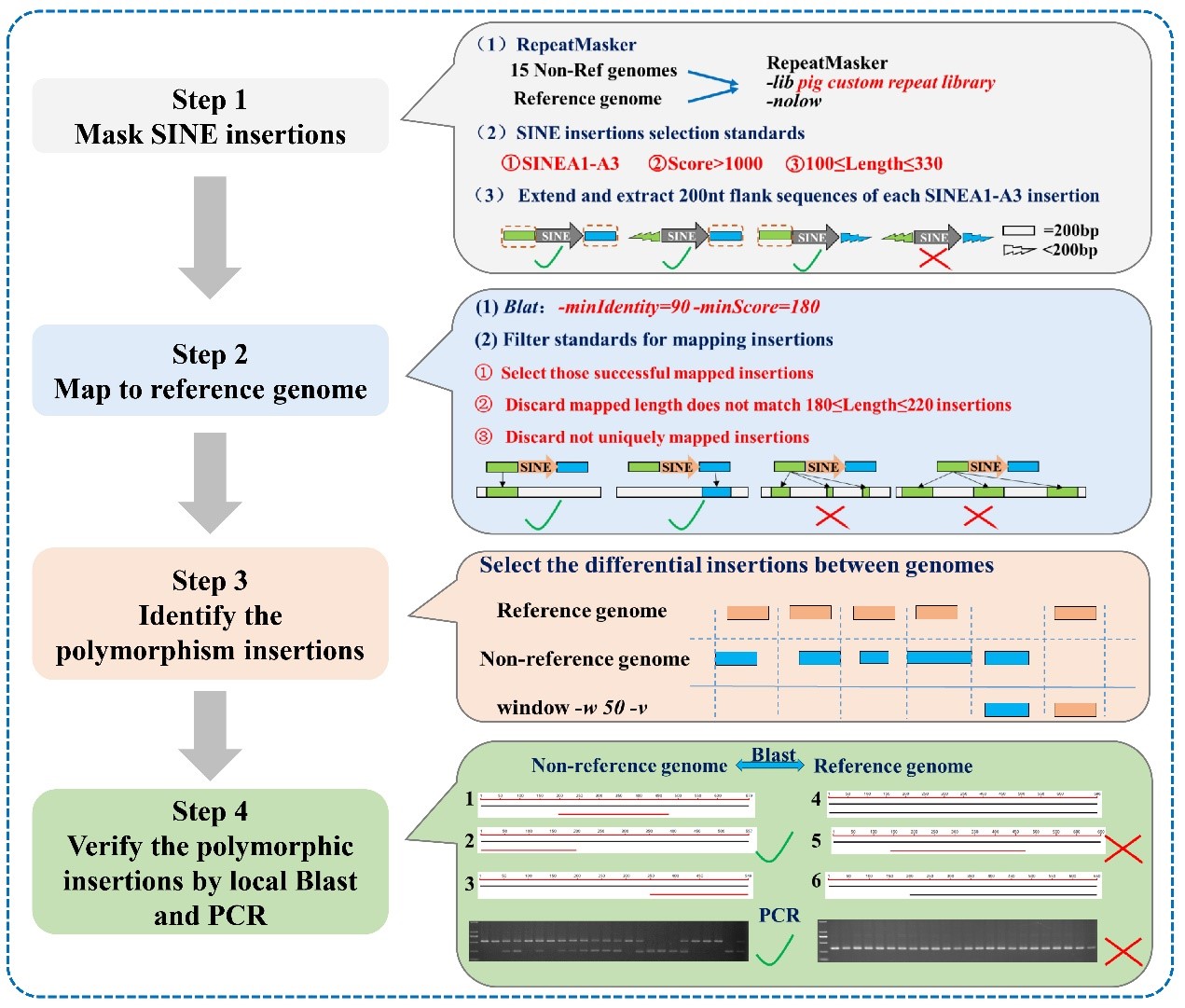
Fig 3. Main steps and methods of SINE RIP annotation.
Reference: Cai Chen; Enrico D'Alessandro; Eduard Murani; Yao Zheng; Domenico Giosa; Naisu Yang; Xiaoyan Wang; Bo Gao; Kui Li; Klaus Wimmers; Chengyi Song; SINE jumping contributes to large-scale polymorphisms in the pig genomes, Mobile DNA, 2021 Jun 28;12(1):17. doi: 10.1186/s13100-021-00246-y. PMID: 34183049; PMCID: PMC8240389.
SINE RIPs mined from short-read genome sequencing data
Accurately genotyping SINE RIPs from large-scale raw sequencing data presents significant challenges. To address this, we developed TypeSINE to genotype SINE RIPs from porcine short-read sequencing data. We assessed its performance using data from 297 domestic pigs and 48 wild pigs, identifying a total of 749,835 SINE RIPs. Among these, 65,917 are common SINE RIPs, with insertion allele frequencies between 5% and 95%, representing only 9% of the total RIPs. About 40% of SINE RIPs are located within the introns of protein-coding genes. A PCR evaluation of 262 loci demonstrated a prediction accuracy exceeding 85%. SINE RIPs are evenly distributed across the genome, with each genome containing approximately 22 common and 260 rare SINE RIPs per megabase. Population genetic analyses using SINE RIPs suggest that Asian and European domestic pigs originated from local wild pigs and were independently domesticated, albeit with subsequent introgression. Additionally, genome-wide association studies identified genomic regions potentially linked to body size. Furthermore, we established PigRIPdb, a database encompassing over 7,500 SINE RIPs, which provides functionalities for browsing, visualization, and searching of RIPs.
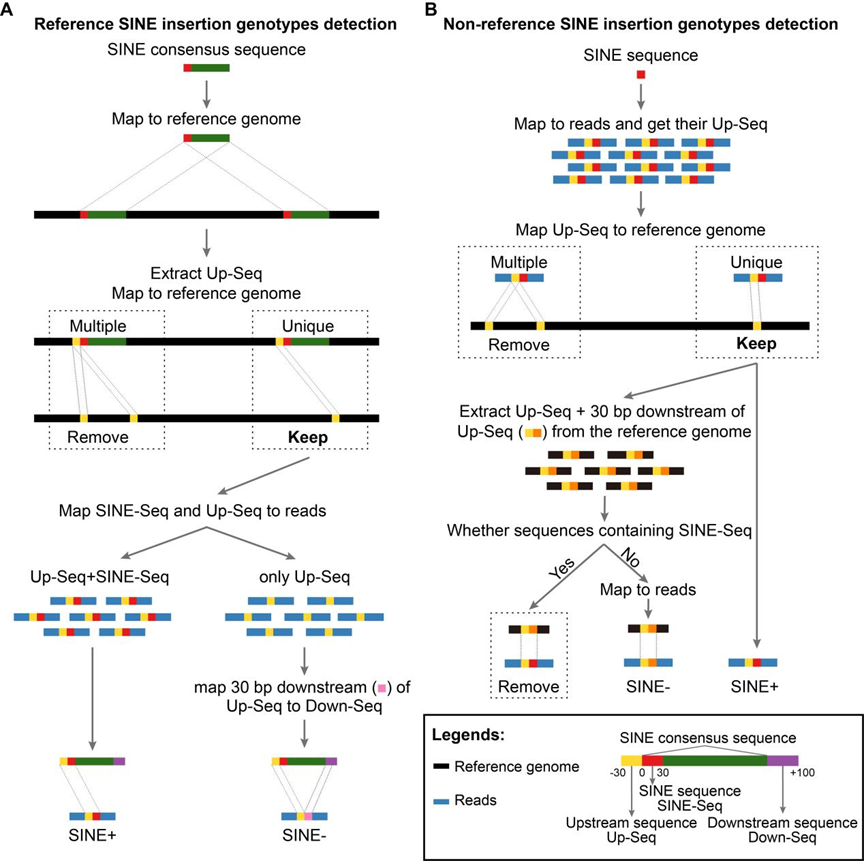
Fig 4. SINE RIP mining protocol. Illustration of the process for detecting SINE retrotransposon insertion polymorphisms (RIPs) using next-generation sequencing data. A. Schematic diagram for reference SINE RIPs detection. B. Schematic diagram for non-reference SINE RIPs detection.